
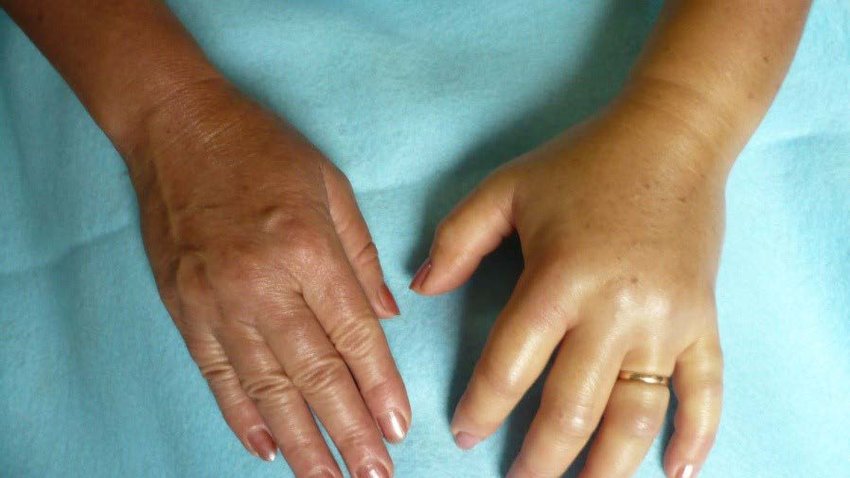
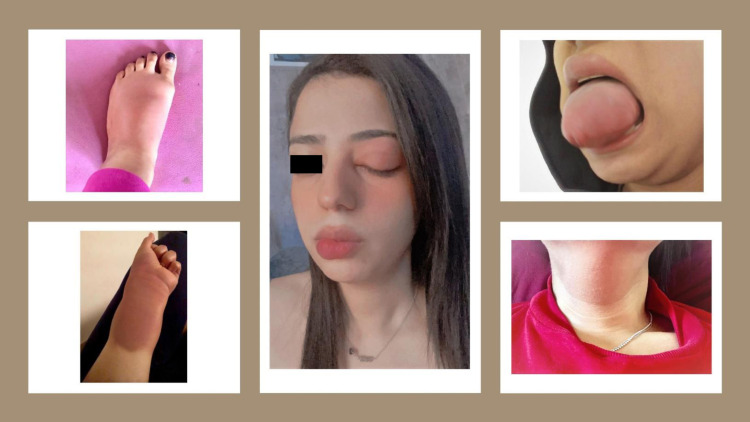
Herediter anjiyoödem ya da kalıtsal anjiyoödem, eller, ayaklar, cinsel organlar, mide, yüz ve/veya boğaz da dahil olmak üzere vücudun çeşitli yerlerinde tekrarlayan şiddetli şişlik (anjiyoödem) ataklarını içeren çok nadir ve potansiyel olarak yaşamı tehdit eden bir genetik durumdur. Hava yolundaki şişme nefes almayı kısıtlayabilir ve ölümcül olabilir.
Epizotlar fiziksel travma veya duygusal stresle tetiklenebilir, ancak şişme genellikle bilinen bir tetikleyici olmadan meydana gelir. Tedavi edilmediğinde, Herediter anjiyoödem atağı genellikle üç gün, bazen daha da uzun sürer ve birçok kişi ayda üç veya daha fazla şişme atağı yaşar. Atakların sıklığı ve şiddeti bireyler arasında, hatta etkilenen aile üyeleri arasında bile önemli ölçüde farklılık gösterir.
Hereditary angioedema kişilerin büyük çoğunluğunda C1-İnhibitör adı verilen plazma proteininde eksikliğe neden olan bir genetik bozukluk vardır. C1-İnhibitörü normal düzeyde olan kişilerde de görülür, ancak diğer genlerdeki genetik bozukluklar anjiyoödeme neden olur.
Kalıtsal anjiyoödem neden olur
Kalıtsal anjiyoödem olan kişilerin büyük çoğunluğunda C1-İnhibitör adı verilen kan proteinini kontrol eden gende bir kusur vardır. Bu kusur, şişmeye neden olan biyokimyasal bir dengesizliğe neden olur. Herediter anjiyoödem ayrıca C1-İnhibitör Eksikliği – Tip I ve Tip II olarak da bilinir. Normal C1-İnhibitörlü Herediter anjiyoödem tanısı alan kişilerde genellikle Tip I ve II’de görülenlere benzer şişlik semptomları görülür.
Herediter anjiyoödem kalıtsaldır ve ebeveynlerinden birinde bu durum varsa çocukların herediter anjiyoödemi devralma şansı %50’dir. Bununla birlikte, aile öyküsünün olmaması kalıtsal anjiyoödem teşhisini dışlamaz çünkü bilimsel raporlar vakalarının %25 kadarının gebelik sırasında C1-İnhibitör geninin spontan mutasyonundan kaynaklandığını göstermektedir. Herediter anjiyoödem olan kişilerin çocukları da bu durumu miras alabilir.
Herediter Anjiyoödem tanısı
Çoğu anjiyoödem veya şişlik vakası Herediter anjiyoödem veya C1-İnhibitör eksikliği DEĞİLDİR.
Herediter anjiyoödem çok nadir görülür ve çoğu insan (tıp uzmanları dahil) bu hastalığa sahip biriyle karşılaşana kadar hastalığa aşina değildir. O zaman bile, semptomlar gelip giderken, Herediter anjiyoödemli kişilerin uzun yıllar boyunca teşhis edilememesi yaygındır. Sık ve şiddetli karın ağrısına kolayca yanlış teşhis konulabilir, hatta bazen gereksiz keşif amaçlı ameliyatlara bile gerek duyulabilir.
Başarılı tedavi ve yönetimi için doğru tanı çok önemlidir. Hereditary angioedema tanısı koymak için kan örneklerinin veya genetik örneklerin laboratuvar analizi gereklidir.
Herediter Anjiyoödem hangi yaşlarda ortaya çıkar?
Herediter anjiyoödem semptomları genellikle yaşamın erken dönemlerinde, çoğunlukla 13 yaşında ortaya çıkar ve ergenlikten sonra şiddeti artabilir. Bu hastalık çok nadir olduğundan, semptomların ilk yaşanmasından sonra doğru bir tanıya ulaşmak on yıl kadar sürebilir.








